
Cellular Mechanisms of Arrhythmia Syndromes
Lee Eckhardt, MD, MS, is the Gary and Marie Weiner Professor of Cardiovascular Medicine Research. A clinical and cellular electrophysiologist, her research and clinical practice focuses on genetic and acquired arrhythmia conditions, with the overarching goal of preventing sudden cardiac death.

Improving Treatment and Preventing Sudden Cardiac Death
The Eckhardt laboratory studies functional genomics, focusing on the cellular mechanisms of inherited and acquired arrhythmia syndromes. Our overarching goal is to find new methods and mechanisms to treat arrhythmias to prevent sudden cardiac death.
The team is particularly interested in understanding the cellular dynamics, channel regulation and cellular microdomains. One area of focus is on potassium inward rectifier channel family Kir2.x and how this relates to arrhythmic disease. We are also interested in the dynamics between structural and signaling proteins and their impact on arrhythmic disease.
The Eckhardt lab also investigates methodology for high-throughput genetic variant characterization, particularly for genes encoding cardiac potassium channels.
A better understanding of these mechanisms could lead to more effective and personalized treatment of inherited and acquired arrhythmia syndromes.
Research Team

Assistant Scientist

Associate Scientist
Corey Anderson's Google Scholar page

Research Associate

Post-doc

Biomedical Engineering PhD Student

Pre-doc

Master’s Student: University of Freiburg, Germany with research component at UW-Madison

Undergraduate Research Assistant
Lab Reunion
Dr. Eckhardt's research team held a reunion at the Memorial Union Terrace.

Former Lab Members
- Molly Melnick, BS, BSN
- Max Millatis, BS
- Mansa Kalluri, BS
- Lexi O’Keffe, MD
- Hannah Waldman, MD
- Seamus McWilliams, BS
- Matthew Kalscheur, MD
- Jason Boynton, PhD
- Jesse Wang, MD, MS
- Hannah VanErt, BSN, MD’21
- Jarrett Warden, MD
- Sara Abozeid
- Igor Bereslavskyy, BS
- Emma Langer, BS
- Kunal Sondhi, BS
- Ravi Vaidyanathan, PhD
- Sam Esch, BS
- Nicholas Fabry, MD
- Cordell Spellman, BS
- Elise McCune, BS
- Walter Stanwood, MS

There are opportunities for motivated individuals in the Eckhardt Lab! We are currently seeking undergraduates, graduate students and postdocs interested in laboratory research in arrhythmia syndromes.
If you are interested in joining the group, please send your CV and a brief description of your research experience and interests to Dr. Eckhardt.
Active Projects
- Loss of Kir2.1
Sudden cardiac death (SCD) is caused by ionic current abnormalities related to inherited or acquired arrhythmia syndromes. One important ionic current associated with various sudden death syndrome phenotypes is IK1.
Genetic loss of IK1 is associated with Andersen-Tawil syndrome (ATS), a disorder comprising ventricular arrhythmia, periodic paralysis and dysmorphic features. KCNJ2 encodes the protein Kir2.1, the dominant potassium inward rectifier in the human ventricle responsible for IK1.
Due to the ubiquitous presence of Kir2.1 in excitable tissue, there is a notable overlap with neurologic symptoms and skeletal muscle abnormalities in ATS patients with loss of normal function mutations in KCNJ2. Ventricular arrhythmia in these patients can include polymorphic ventricular tachycardia and bidirectional tachycardia.
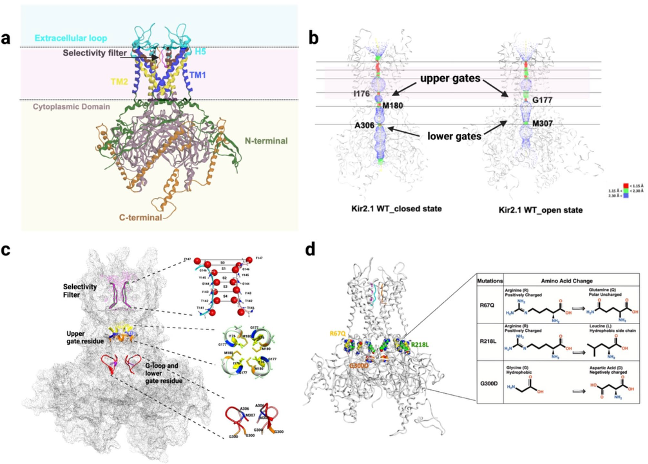
Through the UW Health Inherited Arrhythmia Clinic, the Eckhardt lab has identified several individuals with ATS. We have investigated these mutations from the atomic level to the whole organ level.
A series of patient-specific mutations were studied at the atomic level.
Image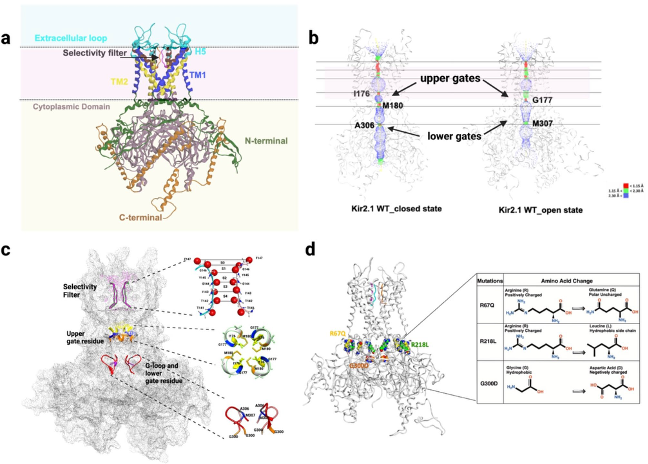
- Rapid Functional Tests
Traditional functional studies and current platforms to determine variant classification are not ideal due to inherent time intensiveness, overall expense and variability in results. We are developing techniques and processes for variant characterization to keep pace with genetic identification.
The K+ channel Kv11.1 is implicated in the SCD syndrome Long QT syndrome 2 (LQT2), and the majority of LQT2 mutations are trafficking-defective. We published a high-throughput method for variant analysis using a bacterial-based solubility assay that can quantitate the degree of protein instability and validated this for characterized Kv11.1 variants.
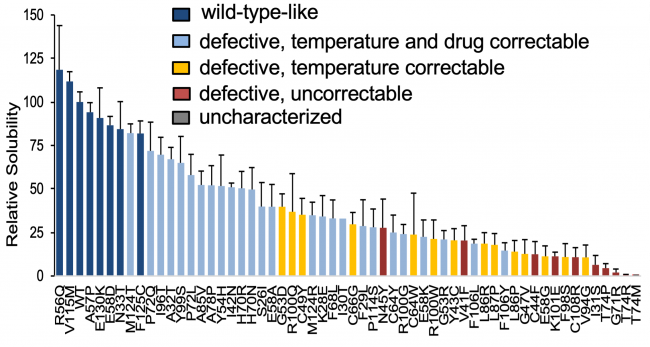
Above: Genetic variant classification by solubility assay. WT-like HERG PAS domain variants have higher relative solubility. More severely trafficking defective variants have lower solubility depending on the severity of abnormality. N≥3/each mutant.
We have also extensively studied genetic variants of the LMNA gene that encodes the nuclear envelope protein lamin a/c, associated with cardiac disease, skeletal myopathy, progeria and lipodystrophy. We have showed in both heterologous cells (both HEK and C2C12) and iPS-cardiomyocytes that lamin a/c cellular aggregation is a major determinant for skeletal and cardiac laminopathies, but not for progeria and lipodystrophy.
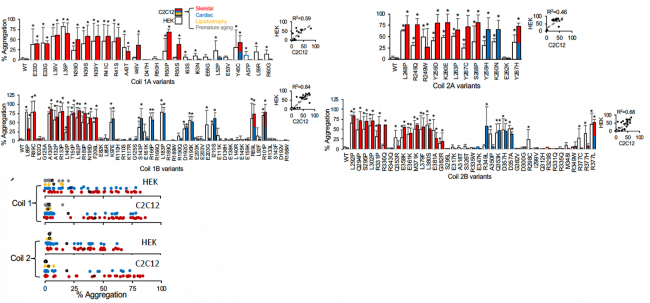
Above: Aggregation of Coil 1A, 1B, 2A, and 2B lamin variants. Red and blue data correlate with skeletal and cardiac disease association. Begin or likely benign variants do not demonstrate aggregation. Summary data is on the left.
This suggests that striated muscle laminopathies are predominantly protein misfolding diseases. Moreover, our high throughput techniques for potassium channels and lamin proteins represent a validated methodology for assessing pathogenicity for myopathic variants of uncertain significance (VUS). This is a major frontier for functional genomics to keep pace with genomic discovery.
- Tools and Models
We use murine transgenic in vivo models for arrhythmia characterization but our main model system is to use human cells to model human disease.
Dominantly we utilize patient-specific induced-pluripotent stem cells that are derived into cardiomyocytes or cardiac fibroblasts and co-cultured on micropatterned PDMS to make biomimetic cardiac microtissues.
For monogenic diseases, we have created “CRISPR corrected” isogenic control iPSC lines or CRISPR modified a control line to contain the mutation. We also use CRISPR gene editing to introduce singular mutations into a non-disease iPSC line, so that we can isolate the mutation-specific effects in an isogenic background. Some of the characterization includes classical current clamp and cellular optical mapping.
- Idiopathic Ventricular Fibrillation
Idiopathic ventricular fibrillation (IVF) is an unrefined diagnosis representing a heterogeneous patient group without a structural or monogenetic definition. IVF treatment is not mechanistic-based due to the lack of experimental patient-models due to several features including a lack of genetic nor structural underpinning for model creation.
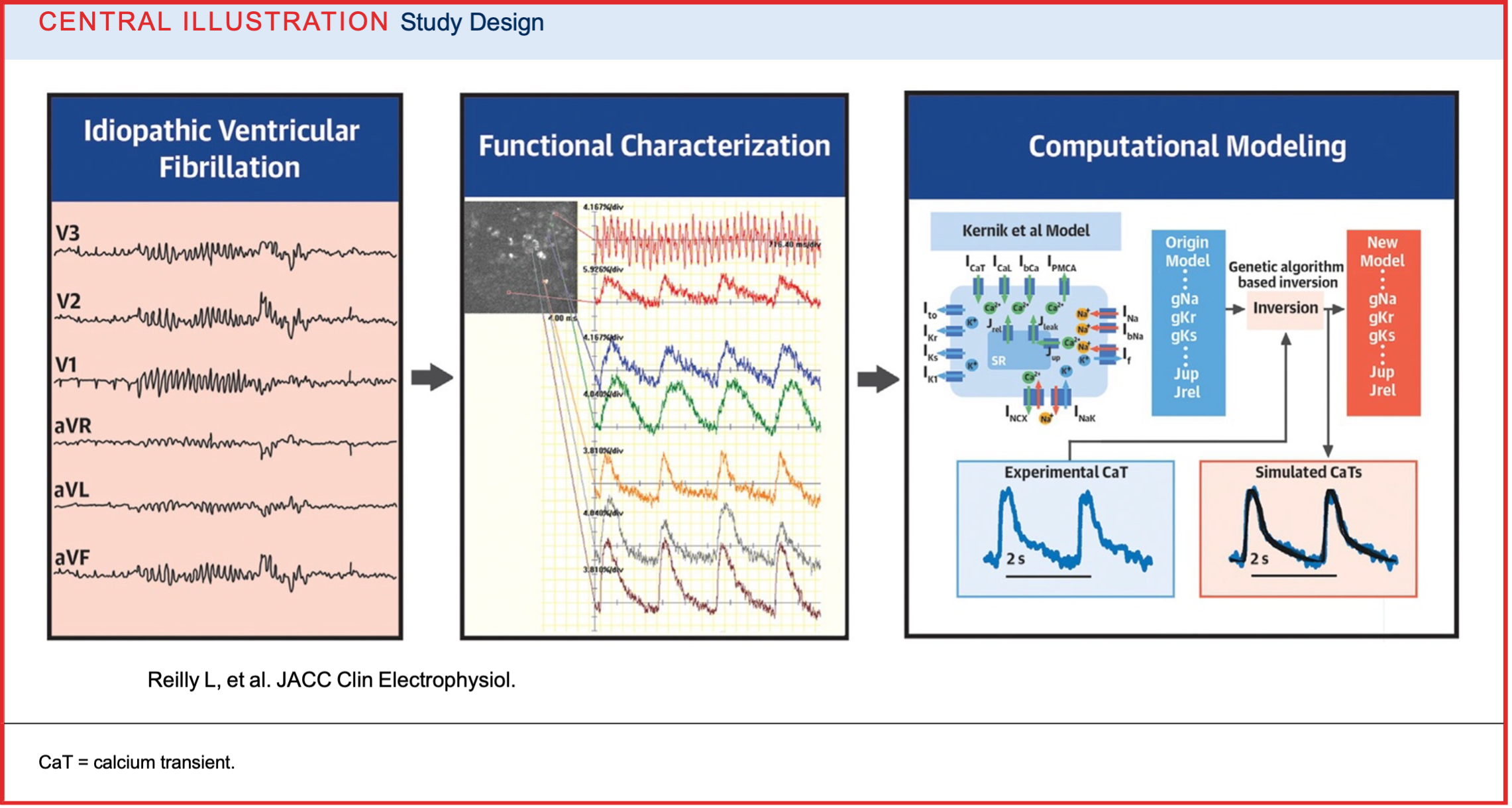
Our lab undertook a new methodologic approach to design mechanistic based treatments for these challenging patients. From a patient with IVF, we created patient-specific induced pluripotent stem cell–derived cardiomyocytes and then integrated electrophysiological optical mapping with computational modeling to characterize the cellular arrhythmic phenotype. This approach flips the traditional paradigm using a biophysically detailed computational model to solve the problem inversely.
Insight into the cellular mechanisms of this patient’s IVF phenotype could also serve as a therapeutic testbed. Our proof of concept paper Central Illustration is depicted below. For further reading: Modeling Idiopathic Ventricular Fibrillation Using iPSC Cardiomyocytes and Computational Approaches: A Proof-of-Concept Study
Image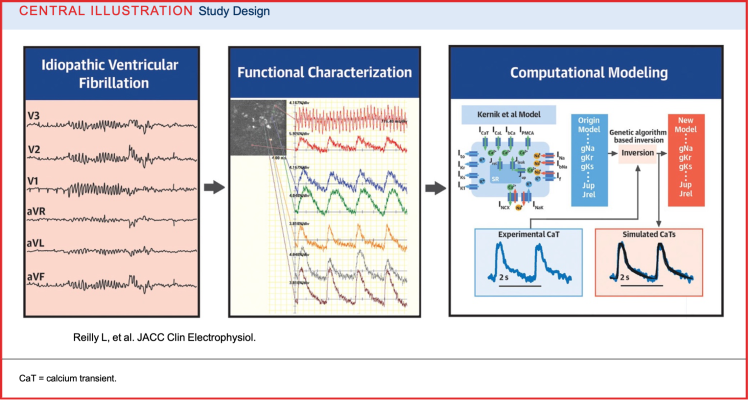
Funding Support
Dr. Eckhardt's funding partners include the Jackson Foundation, the National Institutes of Health, the Wisconsin Alumni Research Foundation, the University of Wisconsin School of Medicine and Public Health, and the UW-Madison Stem Cell and Regenerative Medicine Center
